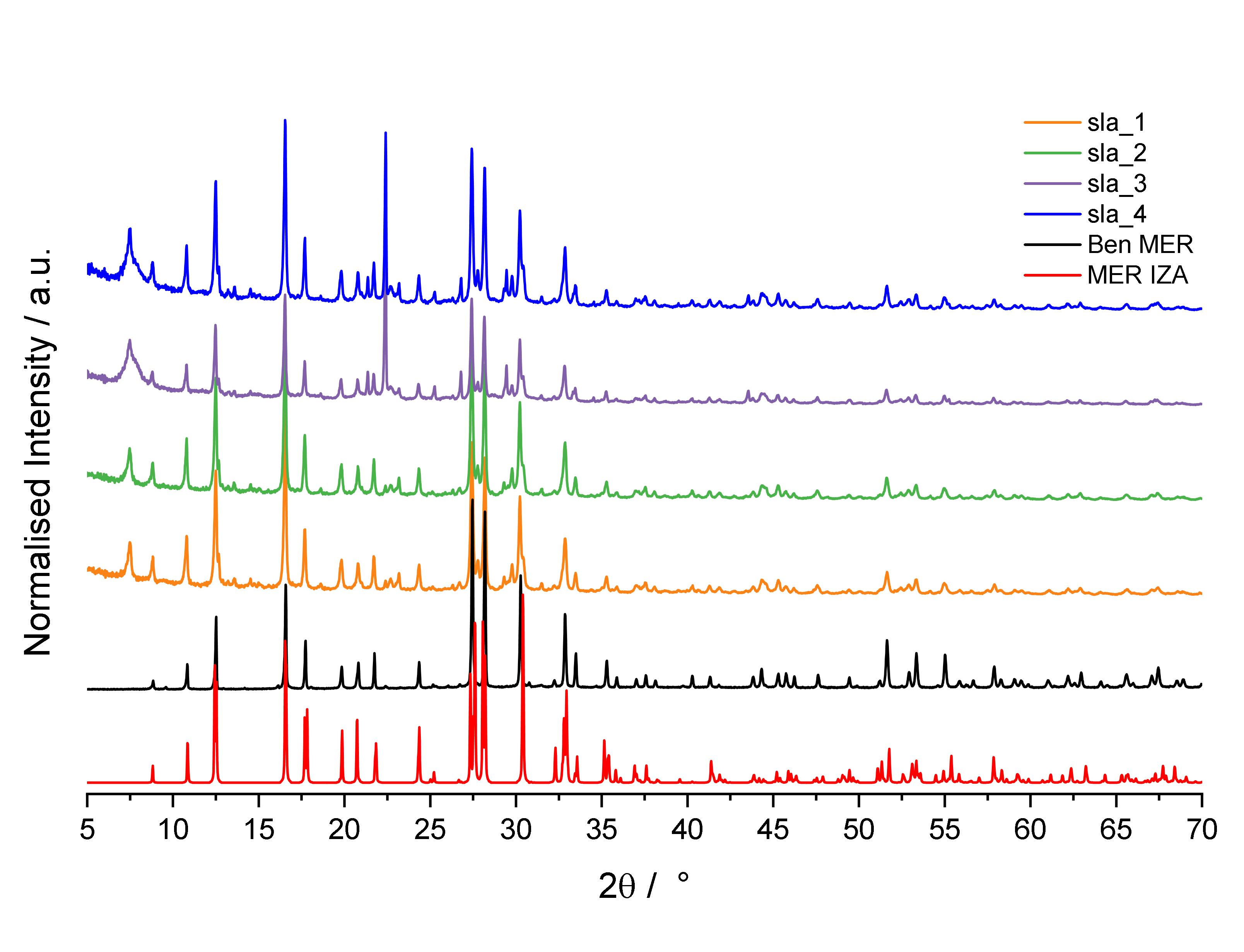
In a way, I am thankful for the partial failure of the syntheses of high silicon: aluminium ratio merlinoites in my research project. Not only because it gives me a much-needed topic for a blog post, but because it presents an interesting conundrum.
The partial failure I am alluding to is that every synthesis I have performed so far, regardless of precursor or method employed, has an impurity. The same impurity. Impurities are a fact of life in chemistry, and for the purposes of our experiments, having a few minor impurities in each sample is insignificant. Gloves get dirty, glassware gets dusty, spatulas are never fully clean – even breathing on your sample can introduce trace impurities. Moreover, the experiments we are interested in are not affected by impurities provided they don’t affect the adsorption of carbon dioxide.
Impurities become interesting, however, when as in this case a series of samples using different apparatus and different materials, prepared on different days, end up with a common impurity. This tells us that it is likely not the random process of physical contamination, but a consequence of our method itself that leads to the impurity we find. Suddenly, finding such an impurity turns from an annoyance to a point of great interest. What is this stuff, and why am I so good at making it by accident?
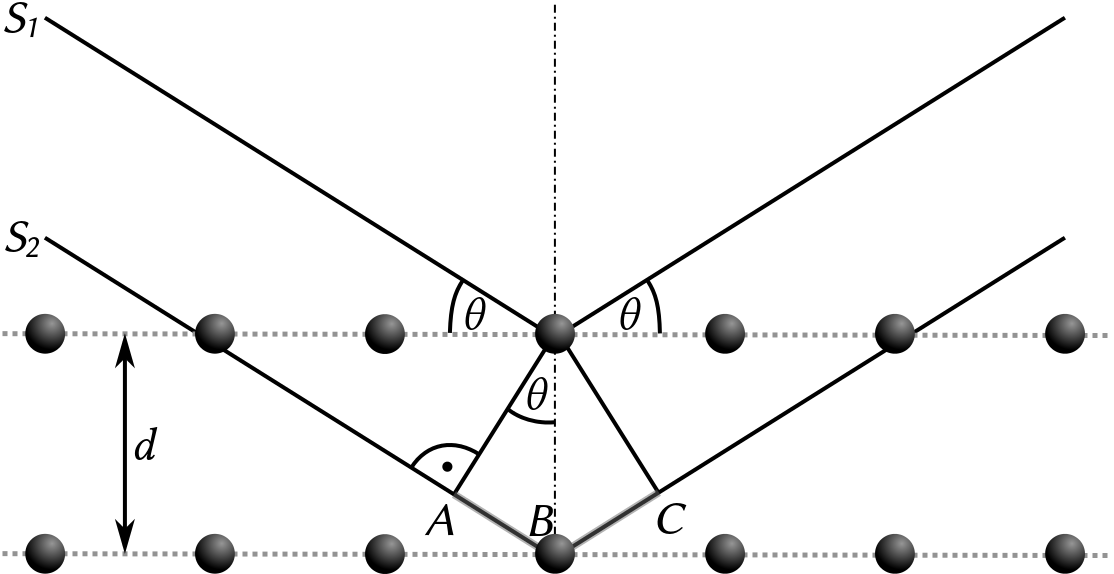
At this point it is probably pertinent to briefly explain the technique by which this persistent impurity has been identified: Powder X-Ray Diffraction (PXRD or just XRD). The technique uses diffraction of X-rays (that is, the spreading of a wave around an obstacle) and subsequent interference of these diffracted waves to elucidate the regular and highly symmetric structures of crystalline solids. This is by virtue of the fact that different spacings of planes (termed Miller planes) within a crystal structure will cause constructive interference of diffracted rays at different angles, related to the plane spacing by Bragg’s Law. Diffracted waves interfere with each other based on the spacing of different sets of planes, which sets their phase difference. Without overcomplicating, essentially by firing X-rays at a sample and recording the angles they are detected (and not detected) at relative to the incident angle, different crystal structures can be very accurately identified.
Numerous databases exist which between them contain the diffraction pattern fingerprints of practically every reported crystal structure. Identifying an impurity should then be as simple as matching one of these patterns to the experimental data. If only. The issue is that superposing two patterns of the intended material and an impurity (or impurities), until it matches the experimental data is somewhat of an idealisation, since it neglects any interactions between X-rays diffracted off the planes in different phases within your sample. Even then, what you identify could well be a new phase not identified before.
So, the scene is set – enter stage left a small but noticeable peak in all my diffraction patterns at 2θ = 7.53°. A peak which matches, as far as I am aware, a grand total of zero known aluminosilicate crystal structures. It may well be something entirely new. How exciting!
Cruelly, science won’t allow me to celebrate my incredible discovery until I have garnered even the faintest idea of what it might be. The peak lies between the expected first peak from MCM-61 and UTD-1: two different zeolites with very different structures and syntheses to the intended product, merlinoite. None of the other peaks expected from either of these frameworks are seen in my experimental XRD patterns. The peak also doesn’t match any found in the XRD patterns of any of the precursors used, and even if it did, this would not explain why it is present in every single sample. It also doesn’t match any that might be expected from the lining of the plates used to run the samples, or anything else which should have been there. The case has seemingly gone cold.
What else could it be? This is the question which haunts my every waking moment. Short of that, it certainly always occupies a part of my mind. More so than worrying about the culprit, I am torn between, on the one hand, my worry that obsessing over this impurity constitutes wandering off on an irrelevant tangent, and on the other hand, my strong conviction that every result is worth investigating.
After all, many of science’s greatest discoveries were made by accident by people who got sidetracked from what they were meant to be doing – X-rays, penicillin, radioactivity, and the cosmic microwave background are all examples. I haven’t quite reached the point of delusion necessary to compare my potential identification of a minor unexpected XRD peak at 7.53° to these era-defining scientific breakthroughs. However, I am curious enough to pursue this unplanned line of enquiry, hopefully leading to my own miniature discovery – even if that discovery is just confirmation that I’m rubbish at making merlinoites.
The peak at 7.53° corresponds to a very widely spaced plane, and as such must arise from a non-microporous phase within the sample. This alone is grounds to dismiss it: such a phase is highly unlikely to be capable of any appreciable gas adsorption, and so will not change the outcome of any adsorption experiments run on the samples. The sensible thing to do is to cast this impurity aside.
However, we all know there is no greater pleasure than studying what you’re not meant to be studying. The feeling that solving this conundrum amounts to nothing but scratching a curious itch of no practical value is a crucial part of its appeal. I look forward to spending many more hours pondering the origins of my now beloved impurity.
I studied Physics and Chemistry, and undertook research in synthetic and analytical inorganic chemistry. The aim of my project was to optimise the carbon dioxide adsorption behaviour (by optimising pore structure changes, gating effects and pore sizes) of merlinoite (zeolite) molecular sieves, for use in carbon capture in industry. This included development of reliable synthetic routes for merlinoites and developing an analytical model for their kinetic adsorption behaviour.
The work will hopefully lead to further work, producing practical results which can be implemented to make gas purification in industrial processes, natural gas upgrading and waste processing more environmentally friendly.
My Leadership in Action programme involved working with Grupo PROMESA on the circular economy by working on their "Recilcando x un Techo" recycling scheme in Mexico City, partnered with Make Sense Americas.
After completing my Laidlaw scholarship, I moved on to read for a DPhil in Chemistry at the University of Oxford.
Outside of studying and research, I have keen interests in writing fiction, amateur rocketry/aeronautics, cookery and philosophy. I play tennis, football and chess.
Recommended Content




Help a fellow scholar by participating in a 10 minute survey: UK Men aged 18-25.

Please sign in
If you are a registered user on Laidlaw Scholars Network, please sign in